Resources
 Part of the Oxford Instruments Group
Part of the Oxford Instruments Group
Expand
Collapse
Part of the Oxford Instruments Group
Application Notes
Author: L. Foglia, M. Wolf and J. Stähler, Fritz-Haber-Institut der Max-Planck-Gesellschaft, Berlin
Published: 01 Apr 2015 · Last updated: 16 Jul 2025
Tags: spectro-detect-solutions-ar, nl-spectroscopy-app, ms-semiconductors-app
Current optoelectronic devices for light harvesting (or emission) are based on heterostructures of semiconducting or metallic compounds with different electronic properties. The efficiency and functionality of the device depends strongly on the rates of charge and energy transfer at the interfaces present in these complex structures and their ratio to other energy dissipation mechanisms. All these processes are transient effects, i.e. they occur for a finite amount of time after the system is photoexcited. Their complete understanding, and thus the ability to engineer increasingly efficient devices, requires spectroscopic techniques for the investigation of even deeply buried interfaces in a time-resolved fashion.
Time-resolved photoemission spectroscopy is suitable to probe interfaces buried under only few nm of material, due to the low mean free path of electrons, Because of the long penetration depth of visible and near IR light in most semiconductors, optical techniques reach the interface. However, linear optical techniques, for example transmission or reflection spectroscopy, are dominated by bulk contributions that bury the interfacial response.
Interfacial specificity can be obtained by taking advantage of wave-mixing phenomena arising when the material interacts with incident light of high intensity via the second order polarization P(2)= ε0 χ (2) E2. Sum frequency generation (SFG), for example, involves two incoming electric fields of frequency ω1 and ω2 and generates an outgoing field of frequency ωSFG= ω1 + ω2. As a consequence of the symmetry constraints on the third-rank tensor χ(2), which is zero in the bulk of media with inversion symmetry, the outgoing field is generated only at the interfaces, where the symmetry is intrinsically broken. Furthermore, P(2) peaks when one of the fields is resonant with an electronic or vibrational transition, making SFG a powerful technique for the spectroscopy of buried interfaces [1]. It is commonly applied to vibrational spectroscopy. Here, a broadband infrared beam, resonant with the vibrational transitions of interest, is up-converted by a second near IR beam, thus obtaining the whole vibrational spectrum of the interface at once.
In order to perform spectroscopy of the electronic states at the interface we instead use a white-light continuum, while keeping the up-converting energy in the nearIR. Combining this probe technique with a third laser beam in a “pump-probe” scheme allows us to achieve time resolution in the femtosecond range. The sample is excited by a first laser pulse, the “pump”, which alters its optical and electronic properties. The pump induced changes affect the polarization and can be monitored by the SFG probe as a function of the time delay between the pump-sample and the probe-sample interaction. To our knowledge a time-resolved electronic SFG approach has been applied only to study liquid interfaces [2].
Figure 1 shows a schematic of the experimental setup. The experiments are performed using a commercial, regeneratively amplified femtosecond (Ti:Sa) laser system (Coherent RegA) working at 40 kHz repetition rate, providing 800 nm light in pulses of 60 fs duration. 50% of the power is used to generate the pump. An optical parametric amplifier (OPA) generates 2.05 eV photons, which are frequency doubled to achieve hνpump = 4.1 eV. The white light supercontinuum (WLC) ranging from 1.78 to 2.48 eV is generated by focusing 5% of the remaining power into a 3 mm thick sapphire crystal. The remaining 800 nm light is used as the upconverting beam.
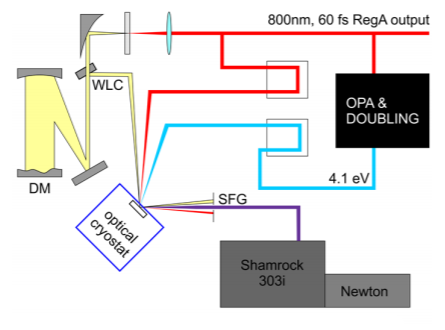
Figure 1 Scheme of the experimental setup showing the generation of the pump via parametric amplification and doubling, generation and compression of the WLC and detection scheme with Shamrock 303i and Newton EMCCD.
To improve the time resolution of the experiment we compress the WLC to 20 fs with a deformable mirror (DM) as described in [3]. The time delay between the WLC and the 800 nm beam as well as the delay between the resulting SFG and the pump pulse is controlled by delay stages that alter the path of the beams in µm steps (3 µm movement 20 fs delay) with an accuracy of 100 nm, i.e. 0.6 fs. The resulting sum frequency spectrum is in the UV region between 3.3 and 4.03 eV (308 to 376 nm) and detected with an Andor Newton EMCCD (DU970P-BV) attached to a Shamrock SR-303i-A spectrometer. For the SFG experiment we use a 300 l/mm grating blazed at 300 nm, which allows measurement of the whole wavelength range on the 1600 pixel detector. The Shamrock is also equipped with a 150 l/mm grating blazed at 500 nm that we use to characterize and compress the WLC. The scattered light of the pump beam is cut by a bandpass filter with a specified transmission band between 314 and 378 nm.
Due to the remaining temporal distribution of the different white light wavelengths and the 800 nm pulse duration of 60 fs, we continuously scan the up-converting 800 nm beam over an interval of ±80 fs with respect to the zero of the cross correlation, thus setting the temporal resolution of the experiment to 160 fs. This scanning range is required to ensure that the delay stage is moving with a constant velocity over the 100 fs cross correlation while the remaining 30 fs before and after it are used for the acceleration of the stage.
The experimental setup is characterized using ZnO. Due to its wurtzite structure, ZnO lacks inversion symmetry and therefore gives a significant bulk contribution to the second order nonlinear effects. The optical band gap of ZnO of 3.4 eV makes it transparent to both the WLC and the 800 nm beam. We expect resonances to occur as final state transitions in the UV. Figure 2 shows one of the first data sets obtained with the setup. In figure 2(a) we depict an SFG spectrum for two sample temperatures. The bottom axis reports the SFG wavelength in nm and the top axis the corresponding energy in eV. We compare the convolution of the WLC spectrum with the RegA output at 800 nm which gives a reference for the expected nonresonant SFG signal, without yet taking into account the system response. The high energy cutoff is due to the bandpass filter closing at 314 nm and the small signature around 300 nm is the residual scattered intensity from the pump beam. Clearly the two curves deviate from each other, indicating an electronic resonance at 361.5 nm (3.43 eV) that grows with decreasing temperature. This means that the electronic SFG is indeed sensitive to electronic transitions in the UV final states of the SFG process.
Figure 2(b) depicts time- and energy-resolved SFG spectra with a false color scale after excitation with the 4.1 eV beam. The response of the system in the absence of the pump beam has been acquired as the background and subtracted. Again, the bottom axis reports the SFG wavelength in nm and the top axis the corresponding energy in eV. The right axis shows the delay between the pump and the SFG probe. Note the split axis: At the bottom it ranges from -1.5 to 4 ps, at the top from 4 to 400 ps. Clearly the SFG intensity changes as a function of pump-probe delay. The right panel shows the one-dimensional time-dependent intensity traces integrated over the regions indicated by the colored rectangles in the 2D plot. Green corresponds to the region between 359.5 and 363 nm, the resonance shown in Fig 2(a). Qualitatively, we observe a fast pump-induced decrease of intensity at this resonance that recovers on a 3 ps timescale and then exhibits long-lived dynamics on timescales longer than the measured interval. The blue region (365 to 370 nm) also shows a sharp decrease at the temporal overlap and then recovers over 300 ps. The assignment of these spectral features is currently under investigation.
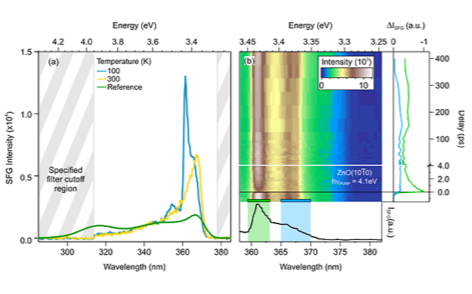
Figure 2 (a) Temperature-dependent SFG signal from bulk ZnO and simulated non-resonant SFG spectrum obtained from the convolution of WLC and 800 nm spectra. Note that the bandpass filter transmits in the region between 314 and 378 nm. (b) Bottom panel: Detail of the SFG spectrum at 100 K reported in (a) in the wavelength region of interest. Top: Background subtracted time- and energy- resolved 2D SFG spectrum measured at 100 K. Right panel: Timedependent traces corresponding to the areas marked by the same color in the 2D plot.
In conclusion we have shown first measurements of electronic resonances and their relaxation dynamics with SFG in a non-centrosymmetric crystal. This data set is the proof of principle that time-resolved electronic sum frequency generation can unveil electronic resonances and their dynamics in solid state compounds. It constitutes the first step towards interface selective time-resolved electronic spectroscopy.
[1] Zhu, X. D., Suhr, H. & Shen, Y. R. Phys. Rev. B. 35, 3047 (1987).
[2] Sekiguchi, K., Yamaguchi, S. & Tahara, T., Femtosecond time-resolved electronic sum-frequency generation spectroscopy: A new method to investigate ultrafast dynamics at liquid interfaces. The Journal of Chemical Physics 128, 114715 (2008).
[3] Wegkamp, D., Brida, D., Bonora, Cerullo, S., Stähler, J., Wolf, M. & Wall, S., Phase retrieval and compression of low-power white-light pulses. Applied Physics Letters 99, 101101 (2011).

© Oxford Instruments 2026
