Resources
 Part of the Oxford Instruments Group
Part of the Oxford Instruments Group
Expand
Collapse
Part of the Oxford Instruments Group
Application Notes
Author: Andor
Published: 01 Apr 2020 · Last updated: 29 Jul 2025
Tags: photonic-solutions-for-chemistry-lc, chemistry-ar
Transient spectroscopy encompasses a powerful set of techniques for probing and characterizing the electronic and structural properties of short-lived excited states (transient states) of photochemically or photophysically relevant molecules. These states are accessed upon absorption of photons and essentially represent higher energy forms of the molecule, differing from the lowest energy ground state in the distribution of electrons and/or nuclear geometry.
For example, it is of fundamental significance to study the excited states of metal-centred complexes and organoporphyrins, both synthetic and naturally occurring. These extensive families of inorganic compounds have wide applicability in technologies such as photomolecular devices and biological probes. It is desirable to probe the exact distribution of charge within the relevant excited state(s) (e.g. on which region of an asymmetric molecule, and in which orbital, does an energetically excited electron reside following photo-excitation). Often one can obtain dynamic information which traces the progression through the states, typically ranging from the microsecond to sub-picosecond regime.
Photochemically interesting molecules typically exhibit efficient absorption of light, and often consist of individual molecular sub-units, in which the electronic influence on each other can instill some very interesting and useful properties into the overall system. These are principally interesting photo-induced properties - the molecular processes that ensue as a result of interaction of the complex with photons, such as emission, electron/energy transfer and photochemistry.
Through intricate examination and characterization of transient excited states the photophysical influences of molecular sub-units can be defined, and one can begin to intelligently design more and more effective and efficient photo-active molecules. For example, these molecules are often capable, in these photosensitized forms, of interacting with or being further influenced by their molecular environment, such as solvents, films, sol-gels or biological molecules, and as such can be designed as effective probes of local molecular environment.
Photo-active molecules can also be used as "building blocks" of much larger molecular structures. For example, metal-centred complexes are used in structurally organized molecular assemblies, yielding supramolecular species capable of performing useful light induced functions. These Photochemical Molecular Devices (PMDs) are providing the bridge between materials and molecules, enabling nanometric machines capable of light-induced functions such as directed electron/energy transfer.
The most intriguing reason underlying use of complexes in PMDs has stemmed directly from nature. Perhaps the most exquisitely effective PMD in existence is the photosynthetic reaction center, enabling ultra-efficient conversion of light into chemical energy, made possible due to a very controlled process of electron transfer following photosensitization.

Whilst the sheer intricacy of such a system renders it impossible to replicate synthetically, it is widely held that it is entirely within our grasp to design artificial PMDs that can successfully duplicate this natural photochemical function. Thus, much research is directed towards the construction of molecular devices capable of performing the photo-induced splitting of water into hydrogen and oxygen. When interfaced with electrodes, these assemblies can convert light into electrical energy and constitute the basis of molecular-driven photovoltaic cells. It is not difficult to imagine the impact that systems based on this technology would have on the world around us, were they to be incorporated into practical devices.
Photo-active molecules are of extensive interest within the fields of biology and medicine. For example, small organic molecules that absorb and fluoresce light with high quantum yield can be employed to specifically tag biological molecules. These fluorophores can be introduced into fixed or living cells and should also be as resistant as possible to photochemical change, so that their emissive properties can be harnessed for as long duration as possible. Molecules such as metal-centred phthalocyanines have been successfully used as photosensitizers in Photo-Dynamic Therapy (PDT). In PDT, the photosensitizer becomes activated by light, but it does not react directly with cells and tissues. Instead, it passes on its energy to molecular oxygen to form a particularly potent species called "singlet oxygen". After it has passed on its energy, the photosensitizer returns to its ground state, where it is available to begin the whole process again.
The combination of a pulsed laser and a high-performance multichannel detector allows for the effective formation and detection of transient species. A battery of complimentary pump-probe techniques are often used in laser spectroscopy laboratories, including:
These time-resolved techniques can be assembled in various optical configurations spanning a wide range of time-resolutions, from millisecond through to femtosecond. Access to such a comprehensive facility, offering flexible and tuneable light sources and covering all temporal orders of magnitude, could be used to generate a complete photophysical profile of the most important molecules, i.e. the complete Jablonski diagram. Indeed, it is becoming increasingly apparent that even the initial, extremely short-lived, singlet excited states can be of intense photochemical interest. For example, the photo-isomerization of rhodopsin, a process fundamental to our vision, is thought to occur from initially formed transient states.
In practice, the complete photophysical mechanism is often constructed through the combined efforts (often collaborative) of many research groups, each having different equipment strengths. For example, one lab might specialize in ultrafast (femtosecond) transient absorption spectroscopy whereas another lab might be equipped for nanosecond TR3, absorption and fluorescence.
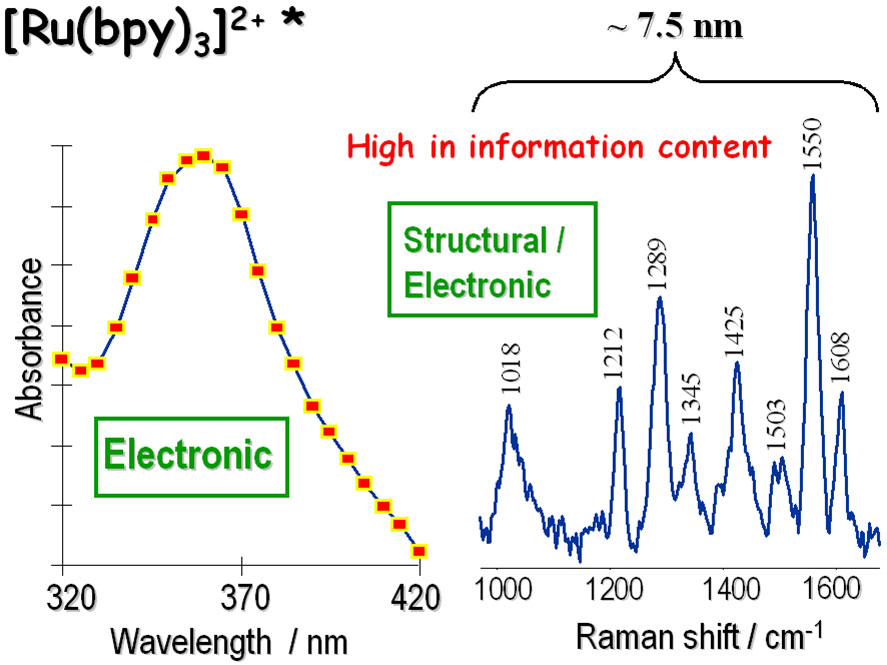
Figure 1 - Comparison of Transient Absorption vs. Transient Resonance Raman spectrum of the 3MLCT triplet excited state of [Ru(bpy)3]2+. A useful Raman spectral region is contained within only ~7.5nm.
Generally, transient absorption and emission spectroscopy provide information on the electronic properties of transient states, and high-quality kinetic profiles of formation and decay can be generated through various stages of a photophysical pathway. The absorption band wavelength is determined by the energy gap of the transition from the probed excited state to a higher excited state. The emission band wavelength is determined from the energy difference between the emitting excited state and the state to which it decays (often the ground state). Whilst the energy gaps between potential well minima of discrete electronic states might seem to be well-defined, the observed absorption/emission bands are often widened by the effects of molecular vibrational broadening and ensemble averaging (the extent of which can depend on the molecule under study and on the temperature of the measured system).
Raman spectroscopy, on the other hand, involves features that have much narrower bandwidths, and much more detailed structural information can be gleaned from this data. Figure 1 shows transient absorption and transient Raman spectra of the triplet excited state of the common metal centred complex [Ru(bpy)3]2+, demonstrating the comparatively rich spectral detail that can be derived from the Raman approach. Furthermore, since the Resonance Raman (RR) effect is also dependent on, and specific to, the absorption transitions of the specific chromophore that is co-energetic with the laser probe wavelength, then high-quality RR data can be generated that affords both structural and electronic information. Time-Resolved Resonance Raman (TR3) Spectroscopy extends this technique into the temporal dimension through introduction of pump-probe capability.
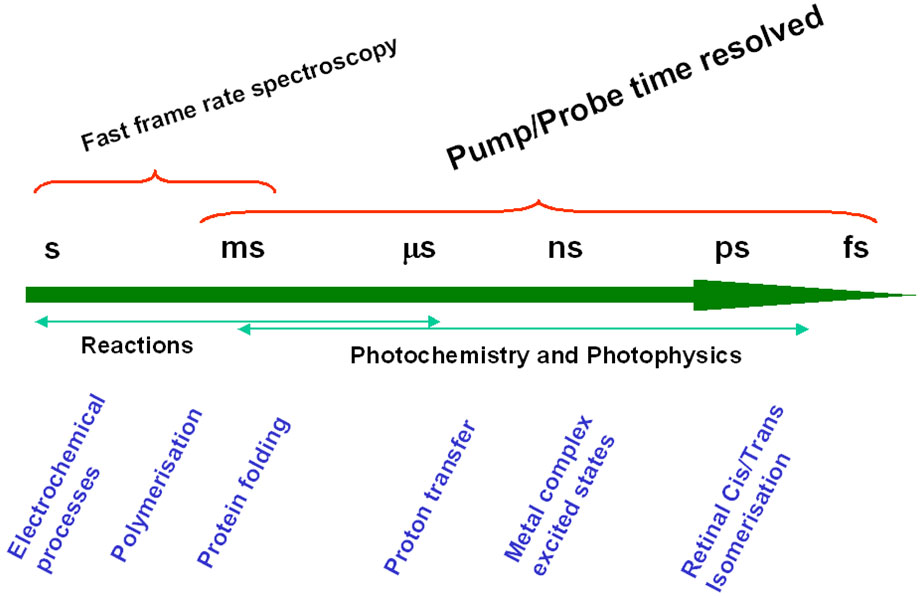
Figure 2 - Fast Frame Rate Spectroscopy vs Pump/Probe Spectroscopy
In the schematic representation of Figure 2, Fast Frame Rate Spectroscopy implies that the signal from the process under study is of sufficient intensity that a kinetic series of short exposures can be employed to record the dynamic process (e.g. a fast photochemical reaction) in "one pass". It also requires that the detector can be readout at sufficiently fast spectral frame rates to enable the kinetics of the process to be followed. With CCD detectors, this is generally possible down to the millisecond temporal regime. The best detector for this type of work is without doubt the state-of-the-art spectroscopic Electron Multiplying CCD (EMCCD), the Andor NewtonEM which allows very fast spectral rates with single photon sensitivity combined with > 90% Quantum Efficiency (QE).
However, in photochemistry and photophysics one is frequently required to study systems with much faster photo-induced pathways and/or insufficient signal strength (e.g. Raman scatter), such that a ‘one-pass’ kinetic study using fast frame rate spectroscopy is impractical. For these systems, pump-probe time-resolved spectroscopy is the answer. In it the sample volume is repeatedly excited and probed by a succession of short laser pulses. The combined "accumulated" signal is collected by the detection system.
Furthermore, this is performed for a series of variable time delays between pump and probe, as illustrated in Figure 3, enabling a kinetic profile to be assembled also. Depending on the specific technique, the probe stage could represent either the opening of an Intensified CCD (ICCD) nanosecond gate, or the firing of a probe laser pulse (or both).
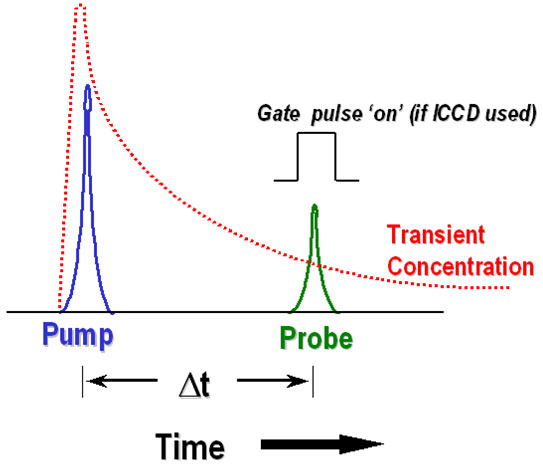
Figure 3 - Schematic representation of pump/probe time-resolved transient
One must also consider the inefficiency of the laser pumping process, that usually only converts a relatively small percentage of the probed sample volume into the excited state manifold – for this reason also, the signal strength is weakened. Importantly, the fastest time-resolutions can be achieved by the pump-probe method, since pulsed laser sources with nanosecond through to femtosecond time-resolution are widely available, and it is generally the temporal overlap (cross-correlation) of pump and probe pulses that ultimately defines the time-resolution of the system. However, in some nanosecond configurations, the temporal resolution of a gated ICCD detector also plays an important part in defining the time-resolution of the set-up. Finally, the time between successive pulses (as dictated by the pulse rep rate) must not exceed the time taken for the molecules under study to decay back to the ground state.
Suffice to say that, compared to typical fluorescence quantum yields, even the enhanced Resonance Raman scatter effect is inherently weak, and when considering also typical low conversion rates to excited state upon pumping, one must almost invariably apply long overall CCD exposures (several minutes are not unusual) during which many pump/probe laser pulse sequences will have occurred, as dictated by the rep rate of the system.
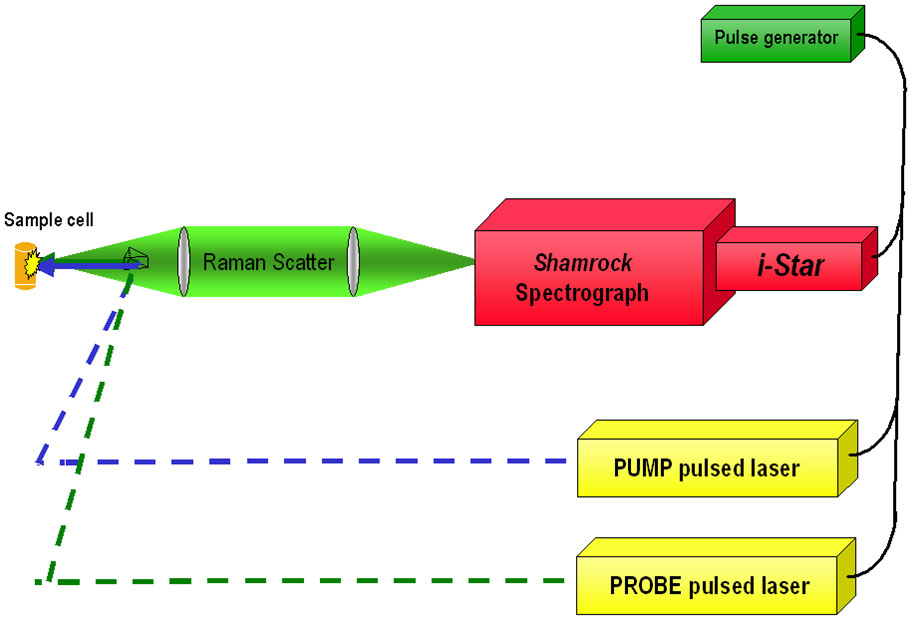
Figure 4 - Schematic diagram of simplified nanosecond-TR3 set-up (backscatter geometry)
In the nanosecond regime, it can be useful to apply a short-gated ICCD Camera (Andor iStar), such that both the extraneous background light and the longer-lived emission of the sample can be "gated out". For example, a 2ns gate can be timed to coincide with the probe laser (of similar or lesser pulse width) to collect the Raman scatter, since Raman is a concerted process. In doing so, we can exclude much of the longer-lived emission that also comes from the sample, induced by the pump or probe lasers - often both.
It is also critical in TR3 to ensure that the probe pulse energy is sufficiently low, in order to certify that it itself does not cause any significant pumping of the sample volume to the excited state. As such, it becomes particularly challenging in TR3 to achieve high Signal-to-Noise ratio (S/N). One solution is to use longer overall exposure times and thus more pump/probe repetitions, but often one has to be careful of sample photo-decomposition from over-exposure. Therefore, the use of high-end detectors with high QE and low noise is recommended to maximize the available signal.
With nanosecond TR3, the option exists to use an Andor iStar ICCD with a Gen III+ intensifier tube (yielding > 40% peak QE), or to use an Andor Newton spectroscopic EMCCD detector with > 90% QE (at the expense of using a non-gated detector). Each type of detector has a zero read noise floor, and as such is single photon sensitive. For picosecond TR3 (i.e. time-resolution with 2 to 3ps cross-correlation between pump/probe pulses), one must use a non-gated CCD. In such cases, the highly flexible and powerful Newton spectroscopic EMCCD represents an excellent choice, as it can be used to maximize SNR whilst keeping overall exposure times short (and therefore the number of pulses limited to less damaging levels) and probe energies low. Newton can also optionally be read out through a conventional low noise amplifier, a mode in which it is configured as a conventional back illuminated CCD.
For multichannel emission spectroscopy in the nanosecond time regime, the iStar ICCD provides a means to probe the emission profile at defined time delays after the pump pulse, and a second probe pulse is not required. For picosecond time-resolution with multichannel detection, some innovative means of optical gating have been developed, such as the "Kerr gate". In this approach, a standard slow-scan spectroscopic CCD can be used to probe the emission. For the Kerr gate approach, a high QE back illuminated detector is favoured to maximize the Signal-to-Shot Noise ratio, because it is often necessary to subtract off the relatively intense longer lived emission(s) (from triplet states for example) that can leak through the optical Kerr gate even when it is "closed".
For multichannel transient absorption spectroscopy, nanosecond time resolution can be achieved by opening an iStar ICCD gate at a defined time-delay after laser or LED excitation, the absorption profile probed by a steady state or stroboscopic white light source (such as Xenon Arc lamp) focused into the probe volume perpendicular to the direction of the pump laser. In the picosecond/femtosecond time regime the transient absorption can be probed by an ultrafast broadband white-light continuum probe pulse, delayed from the pump pulse via an optical delay line.
One of the difficulties in detecting relatively weak transient absorptions is that the excited state population can attenuate the lamp transmission signal intensity by a relatively small amount (ΔT) compared to the shot noise on the signal. This puts the onus on the need to accumulate as high a signal as possible, in order to achieve a high signal-to-shot noise ratio. A high QE CCD means this can be accomplished with a shorter overall exposure time. However, since one must be careful not to saturate the pixels of the CCD, the best way to acquire the signal is in "accumulation mode (spectral stacking)", where multiple back-to-back exposures are summed together per time point.
A new approach to performing rapid accumulation/stacking of transient absorption spectra is to use the Zyla sCMOS detector. Multiple spectral tracks can be configured within the sensor area, and read out at very fast spectral rates of up to 6000 spectra/sec. This may be adopted to simultaneously acquire a reference spectrum and signal spectrum within the same sensor area, representing a highly efficient configuration.
Transient spectroscopy encompasses a powerful set of techniques for probing and characterizing molecular samples.
As the motions of molecules reduce at very low temperatures, only the photochemistry which does not include movement of atoms can proceed. Hence studies at cryogenic temperatures can provide an important insight into protein mechanisms, photochemically interesting molecules and a range of other physical or biological samples.
Cryostats such as the Optistat range, are designed to integrate into existing commercial spectrometers or free-space optics to measure transient absorption spectroscopy at temperatures ranging from 500 Kelvin to as low as 3 Kelvin. Sample environments can be in vacuum or loaded into a tube with a heat-exchange gas to cater for a range of samples from solid state devices to powder, liquid and biological samples.

© Oxford Instruments 2026
