Resources
 Part of the Oxford Instruments Group
Part of the Oxford Instruments Group
Expand
Collapse
Part of the Oxford Instruments Group
Application Notes
Author: R. Richter, D. Wolter, T. Zimmermann, L. Landt, A. Knecht, C. Heidrich, A. Merli, J. Dahl, R. Carlson, T. Möller, T. Rander, Technical University Berlin, Institute of Optics and Atomic Physics
Published: 01 Mar 2013 · Last updated: 16 Jul 2025
Tags: photonic-solutions-for-chemistry-lc
We present photoluminescence spectra of a new class of carbon nanocrystals called diamondoids, and of cyclohexane. For this study, specific isomers of the five smallest members of these perfectly size and shape selectable molecular diamonds have been brought in to the gas phase and irradiated with synchrotron radiation. All the investigated compounds show intrinsic photoluminescence in the ultra violet spectral region.
The spectra are dominated by vibrational fine structure. The origin of these progressions is discussed with the assistance of quantum chemical calculations. The first excited state of adamantane is a molecular orbital with lowered symmetry due to geometrical changes that occur upon excitation. The luminescence of adamantane can be attributed to a transition from this delocalized first excited state into the ground state. Our data indicate that these geometrical changes of the excited state structure is not limited to adamantane and accounts for the luminescence with loosened symmetry selection rules observed in all the other diamondoids.
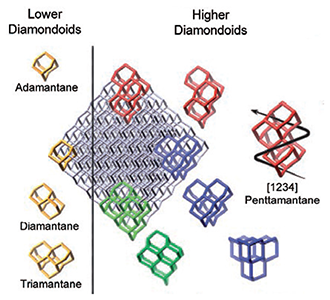
Figure 1: Overview of the structures of selected diamondoids
The size- and shape-dependent optical properties of nanocrystals have attracted large interest in recent years [1, 2]. Such systems are promising candidates for tuning of their optical properties, which is useful for a variety of applications, ranging from new building blocks for optoelectronics to biocompatible, photostable biomarkers [1, 3–5].
In particular, the observation of photoluminescence (PL) in reduced dimensionality semiconductor systems, that otherwise exhibit an indirect bandgap, like Si and C (diamond), opens possibilities to tailor their optical properties to interface with already existent technologies.
Despite numerous studies, the underlying physical processes still are not fully understood [6]. For deeper insight into these, it is desirable that experiments are performed at the idealized conditions that are normally assumed by theory: electrically neutral, isolated systems with well defined sizes and structures. In this work we report PL spectra of a series of perfectly size and shape selected carbon nanocrystals, so called diamondoids, as well as of cyclohexane, which is a subunit of adamantane.
Diamondoids can be seen as cut outs from the diamond lattice. Like in macroscopic diamond, the bonds at the surface are hydrogen passivated [4]. All carbon atoms in pristine diamondoids are sp³-hybridized, resulting in three dimensional structures. The smallest cage-like structure is called adamantane. Corresponding to the number of cages a numerical multiplier (dia-, tria-, tetra-, & pentamantane) is used for naming the polymantanes. Ada-, dia-, and triamantane are called lower diamondoids, since each has only one structural isomer. For the higher diamondoids, the number of isomers increases drastically with the number of cages. Chirality aside, tetramantane has three isomers, pentamantane six and hexamantane already has 24 isomers [7].

Figure 2: Experimental setup at the BESSY II synchrotron radiation facility. 1. beamline, 2. target cell (removed from vacuum chamber), 3. vacuum chamber, 4. vertically mounted Shamrock SR-303i-B spectrometer, 5. Andor iDus DU420A-BU2
For the PL measurements the samples were brought into the gas phase, using a heatable target cell with UV-transparent MgF2-windows. The cell, including the samples, were outgassed at elevated temperatures until the ambient pressure was less than 10-5 mbar. Prior to reaching the sublimation temperature Ts of each respective sample, the target sell was sealed tight.
To minimize thermal broadening of the spectra, the temperature of the target cell during the measurement was kept only slightly above the sublimation temperature Ts of the respective sample. In order to reduce broadening from intramolecular vibrational redistribution (IVR) [8], the excitation energies were chosen as close to Egap as possible, limited by the experimental conditions.
The experiments were performed at the U125/2 10m-NIM beamline at the BESSY II synchrotron radiation facility (Helmholtz-Zentrum Berlin, Germany) The PL spectra were recorded using a Shamrock 303i spectrometer combined with an Andor iDus DU420ABU2.
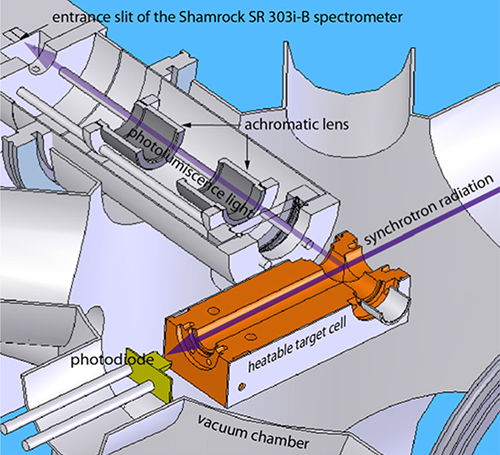
Figure 3. Schematic view of the experimental setup to record the photoluminescence of gas phase diamondoids.
In the following, we discuss the PL spectra, starting with the smallest of the investigated compounds. After cyclohexane, adamantane and the ploymantanes are discussed briefly. to make comparing of the different spectra easier and for the sake of clarity the spectra are shown in one graph and are limited to the three smallest compounds investigated in this work (see Figure 4).
The Stokes shift of the vibronic transition with the highest energy in the emission of cyclohexane (6.57 eV) is 0.47 eV lower than the first allowed transition in absorption. This suggests a major structural change of cyclohexane upon excitation. Cyclohexane, being less rigid then the diamondoids, has two stable conformers.
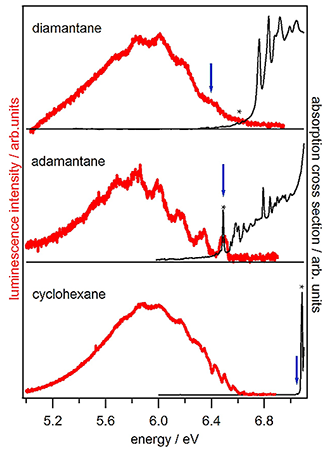
Figure 4. Photoluminescence spectra (red) and absorption spectra (black) of the two smallest members of the diamondoid series and of cyclohexane. The excitation energy is marked with an asterisk and Egap is marked with a blue arrow.
The activation energy to change from low-energy conformer (chair) to the high energy conformer (twistboat) is 10.8 kcal mol-1 [9]. This translates into 0.468 eV and agrees well with the measured shift of 0.47 eV.
Hence we conclude that the very large Stokes shift is likely due to a conformational change upon excitation. Hirayama et al. previously studied the PL of cyclohexane and observed a broad and structurles PL that was attributed to a repulsive singlet state [10] . However, under carefully controlled experimental conditions (low temperature and low excitation energy) vibrational fine structure can be observed. We find no evidence that the initial state of the PL is a repulsive singlet state since the observed broadening can be entirely attributed to IVR.
In contrast to cyclohexane, the highest energy in the emission of adamantane coincides with its first absorption band. This suggests that adamantane is much more stable towards geometrical changes upon excitation.
Our Data show that for the luminescence of adamantane the initial state is the vibrational ground state of the first optical allowed excited state S1. The appearance of the luminescence spectra is determined mainly by the electronic ground state S0, whose potential can be described well by the four vibrational normal modes that are the main contributors to the vibrational progressions. The calculated frequencies of 475 cm-1 (59 meV), 1018 cm-1 (126 meV), 1073 cm-1 (133 meV), and 1373 cm-1 (170 meV) can be assigned to a C-C-C wag (T1), C-C stretch (T2), C--C bend (A1), and a C-H wag (T2) mode respectively [11].
A large number of combinations of these normal modes contribute to the broad background that is apparent in the PL spectrum. Our measurements together with quantum chemical calculations lead us to the conclusion that the first excited state of adamantane is a Rydberg state with lowered symmetry.
Landt et al. have pointed out that for some diamondoids like diamanatane and [121]tetramantane, a smooth absorption onset is observed. Validated by calculations, it has been concluded that in these cases, the lowest transition is symmetry forbidden [1]. These symmetry forbidden transitions cause the absorption cross section near Egap to be very low. Even so, when excited at energies where the absorption cross sections are higher (i.e. allowed states), the initial states for the subsequent emission are the states that were forbidden by symmetry considerations in the absorption. Since relative quantum yields of all samples are of comparable magnitude, we conclude that the symmetry selection rules for dipole transitions, that were found to determine the absorption properties, are less stringent in the case of emission. This can be attributed to the geometrical changes in the excited state-structures after the absorption takes place, reducing the overall symmetry.
We could show that the previously observed, broad and structureless PL of cyclohexane, attributed to a repulsive singlet state, is the result of the experimental parameters. Under carefully controlled experimental conditions (low temperature and low excitation energy) vibrational fine structure can be observed. The comparably high stokes shift in the PL of cyclohexane can be tentatively explained by a conformational change upon excitation.
Adamantane also shows a broad PL spectrum if excited at high energies (>7 eV). This was formerly attributed to the formation of a self trapped exciton. In both cyclohexane and adamantane, the broad and structureless PL that was recorded in previous works is the result of thermal broadening and broadening due to IVR. The luminescence of adamantane, including its vibrational fine structure, can be explained by a transition from the low-symmetry, delocalized first excited (Rydberg) state into the different vibrational normal modes of the ground state.
The photoluminescence properties of the five smallest diamondoids can all be explained by a transition from the vibrational ground state of this first excited state into the different vibrational normal modes of the electronic ground state. The isomerically unique spectral features can be attributed to the different symmetry selection rules of the ground state vibrational normal modes. Our data indicate that the geometrical changes in the excited state is prevalent for all diamondoids. A relaxed excited state with lowered symmetry accounts well for the loosened dipole selection rules observed in the photoluminescence of all the diamondoids.
The broad luminescence in the UV regardless of size, shape, and symmetry implicates that all diamondoids in this size regime may be used as a reliable UV-light sources for technical and biological applications.
We would like to thank the Deutsche Forschungsgemeinschaft DFG for financially supporting this work through grant FOR 1282 MO719/10-1.

© Oxford Instruments 2026
