Resources
 Part of the Oxford Instruments Group
Part of the Oxford Instruments Group
Expand
Collapse
Part of the Oxford Instruments Group
Application Notes
Author: Andor
Published: 10 Feb 2021 · Last updated: 10 Feb 2021
In recent years, single-molecule (SM) fluorescence microscopy techniques have been applied to the study of biomolecules in cultured cells. Extending these techniques to live animal imaging could open new avenues to examine the nanoscale behaviours of signalling molecules under homeostatic control within live tissues and during various developmental or pathogenic stages. SM fluorescence imaging in intact adult animals remains highly challenging, despite previous efforts to track individual biomolecules in embryos and early larval stages of zebrafish and C. elegans nematodes. Although high-contrast imaging techniques such as highly inclined and laminated optical sheet excitation (HILO) or selective plane illumination microscopy have been used successfully for SM in-vivo imaging; advanced labelling methods are still required for site-specific SM imaging.
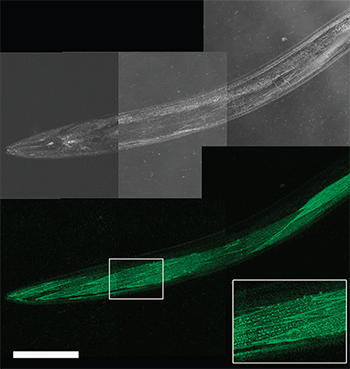
Figure 1. Live worm dCALM confocal imaging of activated CD4-split GFP at the surface of body-wall muscles. Scale bar: 100 mm
Fabien Pinaud and his research team at the University of Southern California have, for the first time, imaged fluorescent biomolecules at the single molecule level in living adult animals. Using far-field fluorescence microscopy and CALM, highly specific, SM sensitivity and nanometre accuracy imaging of membrane proteins within confined tissue regions in intact living C. elegans worms was achieved. CALM was applied to tissue specific imaging and SM tracking of CD4 and voltage-dependent Ca2+ channel (VDCC) membrane proteins in live adult Caenorhabditis elegans (C. elegans). Their results illustrate that SM imaging and tracking by CALM allows a phenotyping of diseases such as muscular dystrophy with sub-resolution precision directly in intact animals.
CALM is a SM technique that allows nanometre precision imaging of individual biomolecules in living cells, independently of their expression levels, at micromolar concentration of fluorescent probes, and without the need to wash excess probes. In CALM, proteins of interest are fused to a dark, non-mature split-GFP that lacks a 16 amino-acid β-strand. They are then fluorescently activated by irreversible binding and complementation with synthetic peptide sequences encoding the missing GFP b-strand (M3 peptides). Microinjection of these M3 peptides allows diffusion throughout the body of animals. They can activate fluorescent signals in engineered muscular and neuronal tissues by targeting and binding irreversibly to cell surface split-GFP fusion proteins. By tuning the concentration of injected M3 peptides, ensemble or continuous SM CALM imaging was performed by direct detection of activated split-GFP, dCALM (Fig 1) or by SM-CALM-spFRET (Fig 2) with targeted near-infrared fluorophores regardless of protein expression levels and of peptide nonspecific binding.
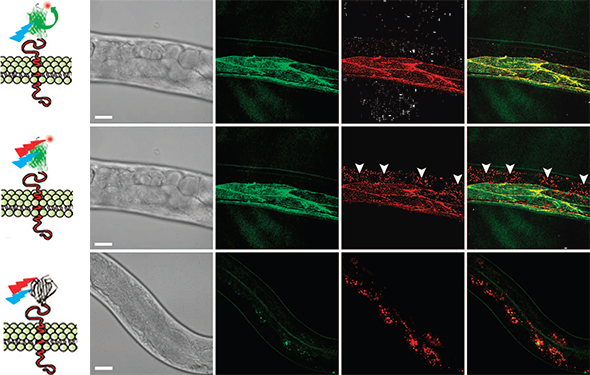
CALM-spFRET confocal imaging of CD4-split GFP in live C. elegans. Single excitation at 488nm allows muscle-specific near-infrared imaging of CD4-split GFP (top) by eliminating interferences from non-specifically bound peptides detected by dual 488 and 647nm excitation (middle, arrowheads). No complementation is observed when injecting worms with the unconjugated A647 dye (bottom). Scale bars: 25 mm.
SM dCALM imaging was performed using 488nm HILO excitation. Under these conditions, activated CD4-split-GFP could be tracked by 2D Gaussian fitting of their point-spread function with a mean localization accuracy of 32±8 nm (Fig. 3). Worms were additionally imaged by SM CALM-spFRET after microinjection of M3-A647 peptides. At the sarcolemma, individual complemented CD4-split-GFPs were identified as diffraction-limited and colocalized diffusing spots that emitted in both GFP and A647 detection channels when excited at 488nm. Single bleaching steps and blinking events where both GFP and A647 fluorescence signals disappeared indicated an effective intramolecular spFRET between CD4-split-GFP and M3-A647 and confirmed to Pinauds research team their ability to specifically target, activate, detect and track individual transmembrane CD4-split-GFP directly in C. elegans.
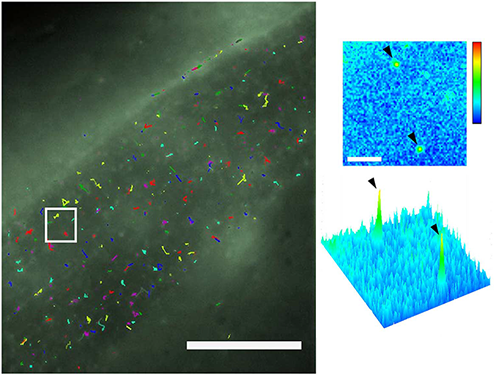
Single-molecule tracking of CD4-split-GFP on muscle cells by dCALM and CALM sp-FRET. (a) Individual activated CD4-split GFP trajectories at the plasma membrane of a dorsal muscle cell. Scale bar: 20mm. (b) Single frame image from the white square showing a 2D and 3D representation of the GFP signal for two individual activated CD4-split GFP (arrowheads). Scale bar: 2 mm
Recently it has been proposed that a dysregulation of VDCC activity in dystrophin mutant C. elegans to be a key factor for the genesis of muscular dystrophy; however, the channels’ location and membrane dynamics have not been studied in living animals. Pinaud and his research team imaged and tracked individual VDCC at the sarcolemma of muscle cells and within neuromuscular synapses of normal and dystrophin-mutant C. elegans, using SM dCALM and HILO excitation. Their study revealed that dystrophin is a load-bearing element and an adaptive tension transducer involved in modulating the nanoscale confinement of VDCC at the sarcolemma in response to changes in muscle tonus.
References
Zhan, H. et al. In vivo single-molecule imaging identifies altered dynamics of calcium channels in ?dystrophin-mutant C. elegans. Nat. Commun. 5:4974 doi: 10.1038/ncomms5974 (2014).

© Oxford Instruments 2026
